About the Lab
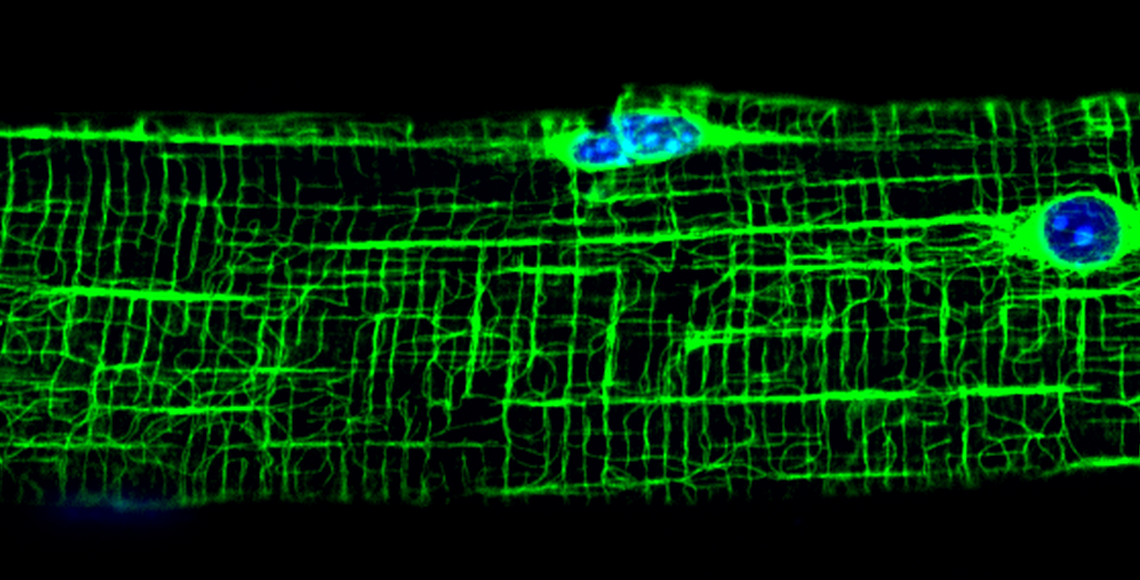
My laboratory primarily studies the structure and cellular function of the dystrophin-glycoprotein complex, which spans the muscle cell plasma membrane (or sarcolemma) and links the cortical actin cytoskeleton with the extracellular matrix. Greater understanding of the physiologic role of the dystrophin-glycoprotein complex is necessary to understand how its absence or abnormality leads to Duchenne muscular dystrophy and forms of human dilated cardiomyopathy. The lab has defined the complete actin-binding region of 400 kDa dystrophin and shown that its homologue utrophin binds actin filaments through a distinct molecular mechanism. Novel methods to visualize the sarcolemmal cytoskeleton without interference from internal structures provided the first evidence that dystrophin functions in vivo to mechanically stabilize γ-actin filaments in costameres. Studies of dystrophin-deficient mice and new animal models generated by the lab have provided insight into the function of costameres in striated muscle and suggest novel links between dystrophin deficiency and alterations in cell signaling, or gene expression manifest by dystrophic muscle. My lab’s unique capability to express biochemical amounts of full-length dystrophin and utrophin has made possible new studies to i) characterize the effects of dystrophy-causing point mutations on dystrophin structure/function, ii) to identify novel associated proteins and iii) to develop new protein-based therapies for dystrophinopathies.
In a completely new line of investigation, my group is working to determine the potentially unique roles of non-muscle actin isoforms in the establishment/maintenance of cell polarity in a variety of tissues. The β- and γ-isoforms of actin distribute to distinct locations within a variety of polarized cell types, including neurons, epithelial cells, and hair cells of the inner ear yet β- and γ-actin differ from each other by only 4 amino acids. Using new isoform-specific reagents and conditional knock-out mouse lines developed during the course of our muscular dystrophy studies, the lab is now working to identify non-overlapping functions of these two highly conserved and widely expressed proteins.