Myosin and Actin | SERCA | RyR l Muscular Dystrophy

Force generation by myosin and actin. Our primary goal is to determine the structural basis for muscle contraction. To do this, we use site directed spectroscopy, transient and steady state enzyme kinetics, and muscle fiber mechanics to measure the coupling between myosin structure, dynamics, and ATPase enzymology. We attach optical and EPR probes to selected sites on both proteins, then use spectroscopy to detect changes in protein structure and dynamics during steady state ATPase cycling, or during transient ligand binding or dissociation steps. Experiments are performed on purified proteins in solution and on intact functioning muscle fibers. The spectroscopic signal allows us to measure changes in structure and dynamics of the labeled proteins. We correlate these changes with the working ATPase cycle and to force generation in muscle.
Spectroscopic probes of muscle contraction form the foundation of our work (described in a recent detailed review Thomas et al., 2009). We have used EPR and optical spectroscopy to show that the myosin head undergoes a disorder-to-order transition during the force-generating weak-to-strong actin binding transition (Baker et al, 1998, LaConte et al., 2003). We have resolved the internal mechanics of this process by measuring the structural dynamics of the myosin catalytic domain using both single and multifrequency EPR (Nesmelov et al., 2008; Agafonov et al., 2008), time-resolved EPR and time-resolved fluorescence to measure interprobe distance distributions by DEER and FRET (Klein et al., 2008; Agafonov et al., 2009). These studies resolved critical questions about how ATP and actin control the structural dynamics of the myosin SH1/SH2 helix, the actin binding interface, the actin binding cleft, and the myosin relay helix. In a recent landmark study, we used a new technique developed in the lab, transient time-resolved FRET (Nesmelov et al., 2011), to investigate the structural kinetics of the myosin relay helix. We found that the helix undergoes a straight-to-bent structural transition during ATP binding. We are currently extending these studies to determine how actin controls the reverse transition, helix straightening, during the powerstroke. EPR experiments using a bifunctional spin label (BSL) are resolving new structural intermediates in the process of force generation (Thompson et al., 2008). BSL attaches rigidly to an a-helix, so it allows us to detect changes in structure and dynamics of an individual helix in the myosin catalytic domain with unprecedented precision and accuracy. We are one of only a small set of labs that use time-resolved phosphorescence anisotropy to measure the structural dynamics of actin. This work shows that actin's cooperative internal dynamics are critical for the actin-myosin interaction (Prochniewicz & Thomas, 2001), and that actin undergoes its own weak-to-strong disorder-to-order transition during active interaction with myosin (Prochniewicz et al., 2004). Different myosin isoforms affect the dynamics of actin in different ways (Prochniewicz et al., 2010)- each actomyosin interaction has its own unique biophysical personality!
The union of spectroscopy and molecular dynamics simulations increases the power of both. We used site-directed spin-labeling (Nelson et al., 2005) and molecular dynamics simulations (Espinoza-Fonseca, et al., 2007; 2008) to show that smooth muscle myosin is activated by a disorder-to-order transition in the phosphorylation domain of the regulatory light chain. Using time-resolved FRET and molecular dynamics simulations, we dissected the structure and dynamics of this domain in the active and inactive state (Kast et al., 2010).
Therapeutic applications. These studies lay the foundation for our understanding of structural dynamics of muscle proteins. We can use this foundation as a cornerstone in setting the development of novel therapies to treat muscle diseases. We are applying spectroscopy to understand the structural basis of muscle degeneration due to aging (Prochniewicz et al., 2007), oxidative stress (Prochniewicz et al., 2008a; 2008b) and muscular dystrophy (Lowe et al., 2006; Prochniewicz et al., 2009).
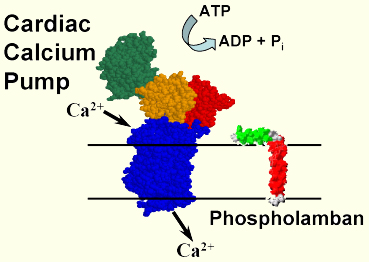
Active calcium transport in the heart by the Ca-ATPase (SERCA) and its regulation by phospholamban (PLB). We are exploring the molecular dynamics of active calcium transport in muscle membranes, using spectroscopic probes of both proteins and lipids. SERCA is an integral membrane protein that uses energy from ATP hydrolysis to pump calcium into the sarcoplasmic reticulum, which relaxes the muscle and provides the calcium gradient needed for the next contraction. By attaching probes to SERCA and performing EPR and phosphorescence spectroscopy, we have shown that active transport involves large-scale movements within the cytoplasmic domain, and that function can be inhibited by reagents (e.g., anesthetics or peptides) that restrict these movements due to aggregation (Mueller et al., 2004).
In the heart SERCA is regulated by phospholamban (PLB), an integral membrane protein that inhibits (and aggregates) SERCA unless PLB is phosphorylated. We used EPR and fluorescence resonance energy transfer (FRET) to show that PLB is in a dynamic equilibrium between monomers and pentamers, but that SERCA binds preferentially to the monomer (Reddy et al, 2003). We used FRET from SERCA to PLB to detect directly the inhibitory interaction and show that relief of PLB inhibition does not require dissociation from SERCA (Mueller et al., 2004). In collaboration with the Veglia lab, we used NMR to determine the atomic structure of the PLB monomer (Zamoon et al., 2003) and to detect the dynamic interaction between SERCA and PLB (Zamoon et al., 2005). We used FRET, in combination with previous EPR and NMR data, to construct a model of the PLB pentamer (Robia et al., 2005). We used peptide synthesis, EPR, and the TOAC spin label to detect directly the peptide backbone dynamics of PLB in lipid bilayers (Karim et al., 2004). We found that phosphorylation induces an order-to-disorder transition in PLB, relieving inhibition without dissociating PLB from SERCA (Karim et al., 2006; Traaseth et al., 2006). Based on this work, we were invited to publish our procedure for synthesis of TOAC-PLB in Nature Protocols (Zhang et al., 2007). We expressed fluorescent fusion derivatives of SERCA and PLB in human cells and measured their interactions by FRET in living cells (Robia et al., 2007; cover article in Circulation Research), and we used similar technology to measure structural changes within SERCA (Winters, et al., 2008).
Toward therapy for heart failure. Both SERCA and PLB have been identified as potential therapeutic targets for treating heart failure (HF), and we are beginning to apply our biophysical approaches to this goal. Celladon, Inc., pointed out that our FRET assay (Mueller et al., 2004) could be used to discover drugs to treat HF, so with their help we developed a high-throughput drug-screening assay. However, we found that this assay, based on steady-state fluorescence, was insufficiently precise, so we are now developing new technology for time-resolved FRET assays in this system, using our newly developed fluorescence instrument that increases the rate of data acquisition by a factor of 100,000 (Muretta et al., 2010), which we have used to develop a fluorescence lifetime plate-reader with Fluorescence Innovations, Inc. (FLPR, see News) Other recent work focuses on designing and testing PLB mutants (PLBM) for use in gene therapy. Previous therapeutic research targeting cardiac calcium cycling has focused on eliminating PLB or upregulating SERCA. Our approach is to introduce a loss-of-function PLBM that competes with WT-PLB and thus activates SERCA. We have shown, along with our collaborators in the Veglia lab, that the dynamics of PLB's cytoplasmic domain correlate with its inhibitory potency and binding affinity to SERCA (Ha et al, 2007, Gustavsson et al., 2011). We are using these structural principles to designing mutants that bind tightly to SERCA while inhibiting less than WT-PLB. This approach has been validated recently in FRET experiments reconstituted proteins (Lockamy al., 2011), and work is currently underway to develop FRET-based competition assays in living cells, using the fluorescence lifetime plate-reader.
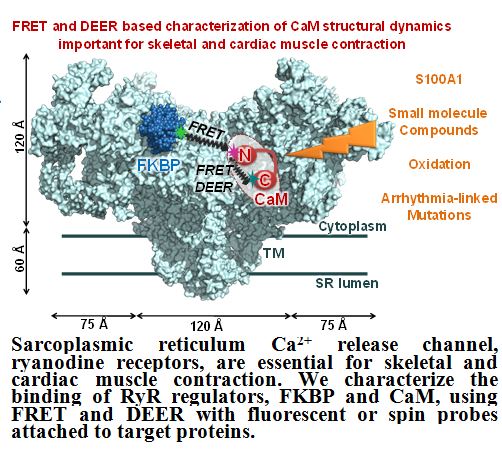
Resolve the structural basis of the sarcoplasmic calcium release channel, ryanodine receptor, modulation under normal and pathophysiological conditions. Embedded in the sarcoplasmic reticulum (SR) membrane of striated muscle, ryanodine receptors (RyR) are responsible for intracellular Ca2+ release that triggers muscle contraction. Dysregulation of skeletal (RyR1) and cardiac (RyR2) isoforms is implicated in the pathology of severe skeletal and cardiac myopathies and neurodegenerative diseases. To maintain normal Ca2+ cycling, RyRs are tightly regulated by endogenous proteins, such as calmodulin (CaM) and FK-506 binding proteins (FKBP) 12.0 and 12.6. Despite clinical importance and potential as a therapeutic target, the FKBP and CaM interaction with RyR is incompletely understood at the molecular level
A primary focus of our lab is to resolve the structural impact of CaM arrhythmia-associated mutants and oxidation on the CaM-RyR interaction. We have characterized the impact of CaM methionine oxidation on CaM-mediated regulation of RyR1 and RyR2 (Balog et al., 2003, Balog et al., 2006). This work is currently extended to explore the structural disruption of these oxidation on CaM and its interaction with RyR1 and RyR2 in native SR membranes or an RyR peptide corresponding to the high-affinity CaM binding domain, using our complementary spectroscopic techniques (DEER and TR-FRET) (McCarthy et al., 2015). This work is essential for understanding the pathological impact of CaM oxidation in heart failure and age-related muscle weakness (sarcopenia).
Our group, in collaboration with the Cornea Lab at UMN and the Bers Lab at U.C. Davis, are using biochemical & molecular biology, fluorescence spectroscopy, and confocal imaging methodologies to help define the structural basis of regulatory interactions within the RyR complex and with RyR regulatory proteins: CaM, FKBP and S100A1 (Fruen et al., 2005, Cornea et al., 2009, Cornea et al., 2010, Mahalingam et al., 2014, Svensson et al., 2014, Oda et al., 2015, Rebbeck et al., 2016). We are currently adapting our basic research for development of high-throughput compatible screening methods for identifying RyR modulators that will be used in the expansion of RyR-targeted therapies for striated muscle diseases.

Structural dynamics of muscular dystrophy. Duchenne (DMD) and Becker Muscular Dystrophies (BMD) are caused by loss of effective mechanical stabilization provided by the protein dystrophin at the sub-sarcolemma. Dystrophin's homolog protein utrophin has shown promise as a therapeutic replacement. Our lab investigates the structural dynamics of the dystrophin-actin and utrophin-actin complexes to understand their function and to help design new constructs for gene and protein therapy. In collaboration with the laboratory of J. Ervasti, we used time-resolved phosphorescence anisotropy (TPA) to show that both dystrophin and utrophin restricted the amplitude but increased the rate of actin flexibility (Prochniewicz et al., 2009, PNAS). This paradoxical combination tunes the dystrophin-actin and utrophin-actin complexes to be strong and resilient (like steel) instead of brittle (like cast iron). Utrophin was much more effective at both restriction of actin rotational amplitude and rate than dystrophin. Current work with other fragments of dystrophin and utrophin, including gene or protein therapy constructs, will determine which structural elements of these proteins are critical in determining the flexibility of actin filaments. In addition, we have elucidated the actin-bound structure of UtrABD1 with high-resolution site-directed spectroscopy and dipolar electron-electron resonance (DEER). UtrABD1 contains tandem calponin homology domains that binds actin in an open conformation through an induced-fit method (Lin et al., 2011, PNAS).
Therapeutic applications: Our lab is pioneering the use of high-resolution time-resolved spectroscopy for the design of therapeutic constructs for Duchenne (DMD) and Becker Muscular Dystrophies (BMD). We have two main approaches:
- Gene therapy. Our goal, in collaboration with the Ervasti laboratory, is to develop a fast and effective screening protocol for promising therapeutic constructs for use in AAV-based gene therapy. We are elucidating the structural and dynamic properties of dystrophin and utrophin as mechanical stabilizers of actin, and we are developing high-throughput FRET assays for detecting the effects of proposed therapeutic constructs on actin flexibility. The goal is to greatly shorten the time required for the initial screening process, greatly decreasing the need for animal trials, which are expensive and slow.
- Drug discovery. Through collaboration with the group of Dawn Lowe, we are investigating the use of small-molecule SERCA activators, discovered in our FRET-based screen in heart failure studies, to decrease intracellular calcium in dystrophic muscles. This approach is validated by recent studies by others showing that upregulation of SERCA in skeletal muscle rescues the dystrophic phenotype in mdx mice. Preliminary trials show that the rate of relaxation in isolated mdx mouse EDL muscle is restored to wildtype values by one of these drug candidates. Our pioneering fluorescence lifetime plate reader is an essential tool in our future drug discovery activities in this field.